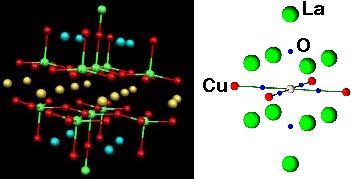
YBa2Cu3O7 and La2CuO4 high-Tc superconductor-clusters used for
density functional calculations
Stripe patterns in underdoped cuprates compatible with magnetic neutron scattering data, E. P. Stoll and P. F. Meier, Phys. Rev. B, 79, 134418
(2009).
Abstract:
In the insulating phase the cuprates exhibit long-ranged antiferromagnetic order which, however, breaks down when they are doped to become metallic. Neutron scattering data show that upon doping the width w of the magnetic structure factors S(q) becomes broader and that their centers shift away from the antiferromagnetic wave vector by an amount delta which grows with increasing doping concentration. The spatial two-point spin correlation can directly be determined from S(q). Using the information available from the position and width of S(q) and assuming random spin phase distributions, we calculated the spin arrangement s/m in direct space. s/m exhibits patterns of vertical and horizontal stripes of finite length such as a two-dimensional nematic fluid. The finite length depends on the width w whereas the distance between two neighboring stripes is proportional to delta. Furthermore, the differences between experimental results obtained by neutron scattering and by nuclear magnetic resonance are discussed.
Ab initio calculations of the electronic structure of cuprates using large scale cluster techniques, S. Renold, C. Bersier, E. P. Stoll and P. F. Meier, arXiv:0711.1606
(Submitted on 10 Nov 2007)
Abstract:
The local electronic structures of La2CuO4, three members of the Yttrium-family (YBa2Cu3O6, YBa2Cu3O7, and YBa2Cu4O8), and to some extent of Nd2CuO4 have been determined using all-electron ab-initio cluster calculations for clusters comprising up to thirteen planar copper atoms associated with their nearest planar and apical oxygen atoms. Spin-polarized calculations in the framework of density functional theory have enabled an estimation of the superexchange couplings J. Electric field gradients at the planar copper sites are determined and their dependence on the occupation of the various atomic orbitals are investigated in detail. The changes of the electronic field gradient and of the occupation of orbitals upon doping are studied and discussed. Furthermore, magnetic hyperfine fields are evaluated and disentangled into on-site and transferred contributions, and the chemical shifts at the copper nucleus are calculated. In general the results are in good agreement with values deduced from experiments except for the value of the chemical shift with applied field perpendicular to the CuO2-plane.
First principles study of local electronic and magnetic properties in pure and electron-doped Nd2CuO4,
C. Bersier, S. Renold, E. P. Stoll, and P. F. Meier, J. Phys.: Condens. Matter, 18,
7481-7495 (2006)
Abstract:
The local electronic structure of Nd2CuO4 is determined from ab-initio cluster calculations in the framework of density functional theory. Spin-polarized calculations with different multiplicities enable a detailed study of the charge and spin density distributions, using clusters that comprise up to 13 copper atoms in the CuO2 plane. Electron doping is simulated by two different approaches and the resulting changes in the local charge distribution are studied in detail and compared to the corresponding changes in hole doped La2CuO4. The electric field gradient (EFG) at the copper nucleus is investigated and good agreement is found with experimental values. In particular a careful study of the various contributions to the EFG exhibits that the drastic reduction of the main component of the EFG in the electron-doped material with respect to La2CuO4 is due to a reduction of the occupancy of the 3d{3z**2-r**2} atomic orbital. Furthermore, the chemical shieldings at the copper nucleus are determined and are compared to results obtained from NMR measurements. The magnetic hyperfine coupling constants are derived from the spin density distribution calculated for different spin multiplicities.
Charge and spin density distributions around Zn impurities in
cuprates,
C. Bersier, S. Renold, E. P. Stoll, and P. F. Meier, Phys. Rev. B, 72, 224514
(2005).
Abstract:
The effect of zinc substitution on the local electronic structure of several cuprates is investigated using first-principles cluster calculations. Clusters comprising 5, 9, and 13 copper atoms in the cuprate plane of La2CuO4, YBa2Cu3O7, and YBa2Cu4O8 are used. Spin polarized calculations with different multiplicities in the framework of the density functional theory enable a detailed study of the changes in the charge and spin density distribution induced by Zn substitution. Furthermore, doping with charge carriers in the above materials is simulated and the resulting changes in the charge distribution are compared to the changes induced by Zn impurities. These differences are then discussed in terms of a phenomenological model related to properties expected from the generic phase diagram. The effects of zinc substitution are rather local and as expected the absolute values of the Mulliken charge at both nearest and next nearest neighbor oxygens to Zn are larger than in the unsubstituted clusters. The calculated electric field gradient at Cu sites that are the nearest neighbor to Zn is found to be somewhat larger than in the unsubstituted cluster whereas that of next nearest neighbors is about 5% smaller. We conclude that the satellite peak in the Cu nuclear quadrupole resonance spectrum occurring upon Zn substitution in YBa2Cu3O7 and YBa2Cu4O8 has its origin at Cu that are next nearest neighbors to Zn.
Suppression of critical properties in doped cuprates, E. P. Stoll, Journal of Physics A: Mathematical
and General, 38 125-132,
2005.
Abstract:
In high-Tc superconductors dopant atoms supply holes or excess electrons.
Electric conduction happens in the neighborhood of dopants within a circle
several lattice constant wide. Percolation of these conducting areas leads
to global conduction. Diffusing d-electrons in these areas can destroy
antiferromagnetism: the Neel temperatures decrease with doping. Based on an
Ising model with antiferromagnetic interactions acting in the part of the
lattice not covered by conducting areas, the specific heat, the staggered
susceptibility and the spin correlation lengths show very broad peaks even for
low dopant concentrations. In doped cuprates, due to the small size granularity
comparable with the sizes of our simulated systems, possible peak height
singularities are always suppressed.
Keywords: Computer modeling and simulation; critical exponents;
percolation and fractals; magnetic impurities.
From next nearest neighbour site percolation to continuum percolation:
Application to high-Tc superconductors, E. P. Stoll, Int. J. Mod. Phys.
C, 15,
321 (2004).
Abstract:
In high-Tc superconductors dopant atoms supply holes or excess electrons, and
electric conduction is established in the neighbourhood of the dopant.
We propose that percolation of these conducting areas leads to
global electric conduction. To investigate these processes numerical procedures
to simulate continuum percolation are developed. The relation between the
concentration of connecting discs and the fraction of the area of the plane
covered by all discs is computed in the whole range between next nearest
neighbour site percolation and continuum percolation. This method is applied
to investigate underdoped copper oxides where small hole or excess electron
concentrations are insufficient to establish electric conduction. The results
of this study are applied to the model of mobile charge carriers which consist
of or are accompanied by diffusing d-electrons. Our investigations show that
with increasing doping the Neel temperature of the antiferromagnetism vanishes
and spin glass states appear in accordance with experiments. Furthermore,
the peaks in the specific heat become very broad. This broadening
due to randomness and loss of translation invariance has been observed
in nuclear magnetic resonance peaks of doped superconducting cuprates.
Keywords: Computer modeling and simulation; percolation and fractals;
fluctuations (superconductivity); mixed state (superconductivity).
Percolation, fractal behavior and high-Tc superconductivity, E. P. Stoll, Journal of
Superconductivity: Incorporating Novel Magnetism, 17, 79 (2004).
Abstract:
Hole suppliers like Sr in doped La2CuO4 are mainly randomly
distributed. Assuming that the holes are dislocated over a few lattice
constants away from the Sr atom, the conducting areas form randomly
distributed circles in the CuO2 layer planes. Conductivity and
also superconductivity can occur only when these circles touch each
other and form percolation clusters. Mobile holes are accompanied by
diffusing d-electrons. Their spin direction is no longer localized on
distinct places, and antiferromagnetism breaks down.
The phase diagram of high-Tc superconductors is discussed
on the basis of a modified continuum percolation model for which the
centers of each circle are located on lattice points. The
inhomogeneities due to the random hole distributions lead to broad peaks
instead of sharp singularities in the static and dynamic response
functions.
Keywords: Superconductivity, impurity concentration, fractal and percolation
fluctuations at superconductivity, numerical simulation.
On the distribution of intrinsic holes in cuprates, E. P. Stoll, P. F. Meier, and
T.A. Claxton, J. Phys.: Condens. Matter, 15
(26 November 2003) 7881-7889
(2003),
Abstract:
First principles density functional calculations on the La2CuO4
crystal, simulated by using the Cu5O26/Cu8La34 cluster have been analysed to
reveal that the Cu 4s orbital is occupied by about 0.5
electrons. Since this may have important consequences on the method of
calculation of the intrinsic hole distribution in cuprates a study of the
frontier orbitals has been made. It is concluded that the Cu 4s
occupancy is a direct result of a charge transfer from the oxygen anions but
does not involve the hole. It is a clear illustration that the hole
distribution cannot be always estimated from the charge density distribution
alone. 60% of the hole remains on the copper while the rest is spread evenly
about the planar oxygen atoms.
Keywords: Density functional theory, local density approximation,
gradient and other corrections; theory, models, and numerical simulation;
Point defects (vacancies, interstitials, color centers, etc.) and defect
clusters.
Charge Distribution in La[2-x]Sr[x]CuO[4], E. P. Stoll, P. F. Meier, and
T.A. Claxton, International
Journal of Modern Physics B, 17,
Nos. 18-20, 3329 (10 August 2003).
Abstract:
The electronic structure of La2CuO4 has been studied by first-principles cluster
calculations before and after doping. Clusters containing up to five planar copper
atoms were investigated using the density functional method. At variance with
band-structure calculations, we have been able to determine the charge and spin
distributions from molecular orbitals expressed as linear combinations of atomic
orbitals localised at the nuclear sites. Doping is achieved by subtracting an
electron from the cluster which, although there is a change of spin state, produces
a charge distribution which is remarkably similar to the charge distribution of the
peripheral charge method which does not involve changes in spin state. The peripheral
charge method is designed to simulate doping by placing a carefully chosen set of
point charges beyond the periphery of the cluster and relies on the supposition that
the hole distribution is closely related to the charge distribution. More importantly
the peripheral charge method enables charge distribution changes to be obtained for
fractional changes in doping. The results show that with fixed nuclear positions,
doping not only depopulates the 2p orbitals but also affects other orbitals.
This redistribution of charges influences the electric field gradients and hyperfine
coupling parameters. The theoretical values for the field gradients are compared to
values derived from experiments.
Keywords: Theory and models; numerical simulation; response to electromagnetic
fields; point defects and defect clusters.
Muon sites and hyperfine fields in La2CuO4,H.U. Suter, E. P. Stoll, and P. F. Meier, Physica
B: Condensed Matter, Volume 326, Issues 1-4, February 2003, Pages 329-332
Abstract:
The local electronic structure of La2CuO4 has been determined using
first-principles cluster procedures. Spin polarized calculations with the
density functional method with generalized gradient corrections to the
correlation functionals have been performed for various clusters containing up
to nine copper atoms. The resulting electric field gradients and magnetic
hyperfine coupling parameters are in good agreement with experiment. We then
have extended the clusters by inclusion of a hydrogen (muon) at various
selected sites and have determined the total energy allowing for lattice
relaxation of neighboring atoms. The hyperfine fields at the muon transferred
from the copper ions have been evaluated. The results are compared with those
at previously suggested muon sites.
Keywords: Cluster calculations; Cuprates; Hyperfine fields
PACS classification codes: 21.60.Gx; 74.72.Dh; 32.10.Fn
Comparison of the Electronic Structures
of La2CuO4, Sr2CuO2Cl2, and Sr2CuO2F2
C. Bersier, E. P. Stoll, P. F. Meier, and
T.A. Claxton, Journal of
Superconductivity: Incorporating Novel Magnetism, 15
(5): 403-408, October 2002
Abstract:
First-principles cluster calculations are reported of the local
electronic structure of the three compounds: La2CuO4,
Sr2CuO2Cl2, and Sr2CuO2F2. The copper 3dx2-y2 and the
planar oxygen 2ps atomic orbitals exhibit a similar degree of
covalency. The out-of-plane orbitals, however, are quite
different with the 3d3z2-r2 atomic orbital lowered significantly in
energy for chlorine and fluorine apical positions.
Keywords:
density functional theory 71.15.Mb, Numerical simulation
78.20.Bh, impurity concentration, 61.72.Ss, Computer
simulation 61.20.Ja, superconductivity 74.25.Jb
Electric field gradients from first-principles and point-ion
calculations E. P. Stoll,
T.A. Claxton, P. F. Meier, Phys. Rev. B,65, 64532-1 (2002).
LANL (Los Alamos Physics
Information Service) Preprint Server, cond-mat/0111371
Abstract:
Point-ion models have been extensively used to determine "hole numbers"
at copper and oxygen sites in high-temperature superconducting cuprate
compounds from measured nuclear quadrupole frequencies. The present
study assesses the reliability of point-ion models to predict electric
field gradients accurately and also the implicit assumption that the values
can be calculated from the "holes" and not the total electronic structure.
First-principles cluster calculations using basis sets centred on the nuclei
have enabled the determination of the charge and spin density distribution
in the CuO2-plane. The contributions to the electric field gradients and
the magnetic hyperfine couplings are analysed in detail. In particular they
are partitioned into regions in an attempt to find a correlation with the
most commonly used point-ion model, the Sternheimer equation which depends
on the two parameters R and gamma. Our most optimistic objective was to find
expressions for these parameters, which would improve our understanding of
them, but although estimates of the R parameter were encouraging the method
used to obtain the gamma parameter indicate that the two parameters may not be
independent. The problem seems to stem from the covalently bonded nature of
the CuO2-planes in these structures which severely questions using the
Sternheimer equation for such crystals, since its derivation is heavily
reliant on the application of perturbation theory to predominantly ionic
structures. Furthermore it is shown that the complementary contributions
of electrons and holes in an isolated ion cannot be applied to estimates
of electric field gradients at copper and oxygen nuclei in cuprates.
First-Principles Calculation of Electric Field Gradients
and Hyperfine Couplings in YBa2Cu3O7 S. Renold,
S. Plibersek, E. P. Stoll,
T.A. Claxton, P. F. Meier, Eur. Phys. J. B
23, 3-15 (2001)
Abstract
The local electronic structure of YBa2Cu3O7 has been calculated using
first-principles cluster methods. Several clusters embedded in an appropriate
background potential have been investigated. The electric field gradients at
the copper and oxygen sites are determined and compared to previous theoretical
calculations and experiments. Spin polarized calculations with different spin
multiplicities have enabled a detailed study of the spin density distribution
to be made and a simultaneous determination of magnetic hyperfine coupling
parameters. The contributions from on-site and transferred hyperfine fields
have been disentangled with the conclusion that the transferred spin densities
essentially are due to nearest neighbour copper ions only with marginal
influence of ions further away. This implies that the variant temperature
dependencies of the planar copper and oxygen NMR spin-lattice relaxation
rates are only compatible with commensurate antiferromagnetic correlations.
The theoretical hyperfine parameters are compared with those derived from
experimental data.
First Principles Investigation of Local Distortions in Doped La2CuO4
Samo Plibersek, E. P. Stoll, and P. F. Meier, Journal of
Superconductivity: Incorporating Novel Magnetism, 13, 921-923 (2000).
Influence of Spin-Orbit Couplings to Nuclear Spin-Lattice
Relaxation Rates in Sr doped La2CuO4
E. P. Stoll,
S. Plibersek, S. Renold, T. A. Claxton, and P. F. Meier, Journal of
Superconductivity: Incorporating Novel Magnetism, 13, 971-975 (2000).
The electronic structure of Sr-doped La2CuO4 has been investigated by
means of cluster calculations. The results indicate changes in the energies of
the molecular orbitals when a copper atom is in the neighbourhood of a Sr;
in particular the spin-orbit coupling is altered. It is shown that the Cu
nuclear spin-lattice relaxation time depends crucially on this coupling when
measured along the direction perpendicular to the CuO2 planes. It is
argued that the results presented here are compatible with recent experimental
observations.
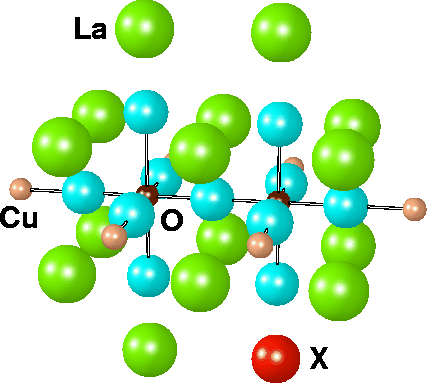 The Cu2O11/Cu6XLa15 cluster. X is either La3+ or the dopant Sr2+. For the
two central Cu atoms (dark) at the L- and S- sites and for 11 O atoms an all
electron calculation is performed. For the other 6 Cu (bright), X and 15 La
atoms pseudopotentials are used.
The Cu2O11/Cu6XLa15 cluster. X is either La3+ or the dopant Sr2+. For the
two central Cu atoms (dark) at the L- and S- sites and for 11 O atoms an all
electron calculation is performed. For the other 6 Cu (bright), X and 15 La
atoms pseudopotentials are used.
Cluster Calculations of Hyperfine Interactions in Copper Oxides
P.F. Meier,
T. A. Claxton, P.
Huesser, S. Plibersek, and E. P. Stoll,
Z. Naturforschung 55 a, 247-255 (2000).
First-Principles Calculations of Hyperfine Interactions in La2CuO4
P.
Huesser, H.U. Suter, E. P. Stoll, and
P.F. Meier, Phys. Rev. B, 61, 1567-1579 (2000).
Cluster Calculations of the Hyperfine Properties of Copper Compounds, H.U. Suter, P.
Huesser, E.
P. Stoll, S. Schafroth, and
P.F. Meier,
Hyperfine Interactions, 120/121, 137-140 (1999).
A User Project: First-Principles Calculation of Electric-Field Gradients
at the Cu Sites in the High Temperature Superconductor
YBa2Cu3O7
P.
Huesser, S. Schafroth,
E. Stoll, H.U. Suter, and
P.F. Meier, CrosSCutS,
7, #2, 15 (1998)
.
First-Principles Calculation of Electric-Field Gradients
at the Cu Sites in YBa2Cu3O7
P.
Huesser,
E.P. Stoll, H.U. Suter, and
P.F. Meier, Physica C,
294, 217-224 (1998).
First-Principles Calculation of Electric-Field Gradients
at the Cu Sites in YBa2Cu3O7
P.
Huesser, S. Schafroth,
E. Stoll, H.U. Suter,
P.F. Meier,
Helv. Phys. Acta, 70, Separanda 2, S25-S26 (1997).
First-Principles Calculation of Hyperfine Interactions in La2CuO4
P.
Huesser, H.U. Suter,
E.P. Stoll,
and
P.F. Meier.
Presented at the
Swiss Workshops on Superconductivity and Novel Metals, Les Diablerets,
September 27-29, 1999.
We present the results of first-principles cluster calculations of the
electronic structure of La2CuO4. Several clusters containing up to nine
copper atoms embedded in a background potential were investigated.
Spin-polarized calculations were performed both at the Hartree-Fock level
and with density functional methods. The dependence of the electric-field
gradients at the Cu and the O sites on the cluster size is studied and the
results are compared to experiments. The magnetic hyperfine coupling
parameters are carefully examined. Special attention is given to a
quantitative determination of on-site and transferred hyperfine fields.
We provide a detailed analysis that compares the hyperfine fields obtained
for various cluster sizes with results from additional calculations of
spin states with different multiplicities. From this we conclude that
hyperfine couplings are mainly transferred from nearest neighbor Cu2+
ions and that contributions from further distant neighbors are marginal.
The mechanisms giving rise to transfer of spin density are worked out.
Assuming conventional values for the spin-orbit coupling, the total
calculated hyperfine interaction parameters are compared with those
derived from experiments.
Theoretical Determination of the Electrical Field Gradients at the
Cu nuclei in high-Tc superconductors
P.
Huesser,
E.P. Stoll, H.U. Suter, and
P.F. Meier.
Presented at the
Swiss Workshops on Superconductivity and Novel Metals, Les Diablerets,
September 30 - October 2, 1996 and September 29 - October 1, 1997.
Nuclear magnetic and quadrupole spectroscopy has provided a considerable
amount of data on both static and dynamic properties of high-temperature
superconductors [1] In particular, the electric-field gradient
(EFG) has been determined with great accuracy for a variety of nuclei.
Using first-principles cluster procedures we have investigated these EFG's
at the Cu sites in YBa2Cu3O7. For the planar Cu, a large cluster
comprising 74 atoms was studied with both Hartree-Fock (HF) and density functional
methods. The latter give a highest occupied molecular orbital (HOMO) which
consists of an antibonding hybridization between the d[x**2-y**2] of Cu(2)
and the p[x] and p[y] orbitals of the neighboring four planar O atoms.
The LDA [2] and GGA [3] results differ only in
details. The HF method, however, produces a HOMO with Cu s and O s and
p[z] orbitals. For all three methods, the EFG's at the Cu(2) site are
closer to the experimental value [4] han those obtained
with other theoretical approaches [5,6]. The electronic
structure revealed by the occupied molecular orbitals and the origin of the
EFG is analyzed and discussed.
The pressure dependence of the EFG at the Cu(2) site is
determined varying the lattice parameters [7]. This
simulation of the influence of hydrostatic pressure is in good
agreement with NQR experiments [8].
References:
[1] For a review, see D. Brinkmann and M. Mali, in ``NMR Basic Principles
and Progress'', (Springer, Heidelberg, 1994), Vol 31, p 171.
[2] S. H. Vosko, L. Wilk and M. Nussair,
Can. J. Phys. 58, 1200 (1980).
[3] A. D. Becke, Phys. Rev. A38, 3098 (1988).
[4] C. H. Pennington, D. J. Durand, C. P. Slichter, J. P. Rice, E. D.
Bukowski, and D. M. Ginsberg, Phys. Rev. B39, 2902 (1989).
[5] N. Sahoo, S. Markert, T. P. Das, and K. Nagamine, Phys. Rev. B41,
220 (1990).
[6] K. Schwarz, C. Ambrosch-Draxl, and P. Blaha, Phys. Rev. B42,
2051 (1990).
[7] J.D. Jorgensen, S. Pei, P. Lightfoot, D.G. Hinks, B.W. Veal, B. Dabrowski,
A.P. Paulikas, and R. Kleb, Physica C, 171, 93 (1990).
[8] K. Mueller, M. Mali, J. Roos, and D. Brinkmann,
Physica C, 162-164, 173 (1989).
View into the YBa2Cu3O7 (YBCO) high-Tc superconductor
For more information see:
Doping high-Tc superconductors with oxygen and metallic
atoms: a molecular dynamics study
Erich Stoll,
Christian
Stern,
Johannes
Singer, and
Peter
Stucki.
Poster presented at the
Swiss Workshop on Superconductivity and Novel Metals, Les Diablerets,
September 30 - October 2, 1996.
The magnetic and superconducting properties of high-Tc superconductors
depend very heavily on the oxygen content of these compounds. This
content can be controlled by the pressure of the ambient oxygen atmosphere
depending on the temperature of synthesis and sample annealing.
To simulate this experimental behavior by the Interactive
Molecular Dynamics System (IMDS) package, an array of YBCO unit-cells
is investigated based on a simplified net interaction with repulsive and
attractive forces of the type V(dij) = 4 [|E|(S/dij)**12-E(S/dij)**6],
where S and E are tunable parameters and dij denotes the interatomic
distance between the atoms i and j. A proper choice of these parameters
allows the identification and reproduction of known stable YBCO structures.
Based on this work, we are able to study the influence of new metal
and oxygen dopant atoms and defects, i.e. the system stability,
and by annealing to zero temperature the reconstruction of the atomic
structure and the appropriate ground-state energy. Therefore, this method
allows an in-depth investigation of processes involved in the synthesis of new
structural compounds and of the chemical and structural stability of
new compounds, and it reveals information for the understanding of
structural phase changes.
The simulation using the Interactive
Molecular Dynamics System package gives not only a sophisticated online
visualization of the processes involved in the formation of a certain
structure, but also provides tools for interactive manipulation and control
of these largely nonunderstood chemical and physical processes.
Doping high-Tc superconductors with oxygen and metallic
atoms: a molecular dynamics study
Erich Stoll,
Christian
Stern,
Johannes
Singer, and
Peter
Stucki, Journal
of Materials Research, 12(11),
2901 (1997).